
This is the mapper pipeline from the Sequana projet
| Overview: | This is a simple pipeline to map several FastQ files onto a reference using different mappers/aligners |
|---|---|
| Input: | A set of FastQ files (illumina, pacbio, etc). |
| Output: | A set of BAM files (and/or bigwig) and HTML report |
| Status: | Production |
| Documentation: | This README file, and https://sequana.readthedocs.io |
| Citation: | Cokelaer et al, (2017), 'Sequana': a Set of Snakemake NGS pipelines, Journal of Open Source Software, 2(16), 352, JOSS DOI https://doi:10.21105/joss.00352 |
If you already have all requirements, you can install the packages using pip:
pip install sequana_mapper --upgrade
You will need third-party software such as fastqc. Please see below for details.
This command will scan all files ending in .fastq.gz found in the local directory, create a directory called mapper/ where a snakemake pipeline can be executed.:
sequana_mapper --input-directory DATAPATH --mapper bwa --create-bigwig sequana_mapper --input-directory DATAPATH --mapper bwa --do-coverage
This creates a directory with the pipeline and configuration file. You will then need to execute the pipeline:
cd mapper sh mapper.sh # for a local run
This launch a snakemake pipeline. See .sequana/profile/config.yaml to tune the behaviour of Snakemake.
With apptainer, initiate the working directory as follows:
sequana_rnaseq --use-apptainer
Images are downloaded in the working directory but you can store them in a directory globally (e.g.):
sequana_rnaseq --use-apptainer --apptainer-prefix ~/.sequana/apptainers
and then:
cd rnaseq sh rnaseq.sh
This pipelines requires the following executable(s):
- bamtools
- bwa
- multiqc
- sequana_coverage
- minimap2
- bowtie2
- deeptools
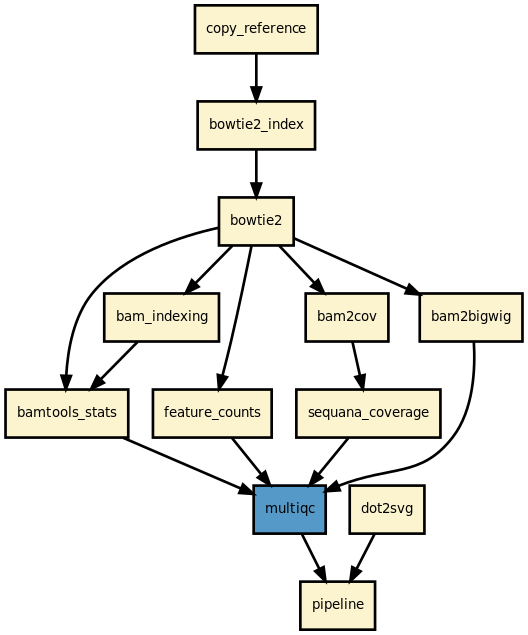
This pipeline runs mapper in parallel on the input fastq files (paired or not). A brief sequana summary report is also produced. When using --pacbio option, -x map-pb options is automatically added to the config.yaml file and the readtag is set to None.
The BAM files are filtered to remove unmapped reads to keep BAM files to minimal size. However, the multiqc and statistics to be found in {sample}/bamtools_stats/ includes mapped and unmapped reads information. Each BAM file is stored in a directory named after the sample.
Here is the latest documented configuration file to be used with the pipeline. Each rule used in the pipeline may have a section in the configuration file.
| Version | Description |
|---|---|
| 1.3.1 |
|
| 1.3.0 |
|
| 1.2.1 |
|
| 1.2.0 |
|
| 1.1.0 |
|
| 1.0.0 |
|
| 0.12.0 |
|
| 0.11.1 |
|
| 0.11.0 |
|
| 0.10.1 |
|
| 0.10.0 |
|
| 0.9.0 |
|
| 0.8.13 |
|
| 0.8.12 |
|
| 0.8.11 |
|
| 0.8.10 |
|
| 0.8.9 |
|
| 0.8.8 |
|
| 0.8.7 |
|
| 0.8.6 |
|
| 0.8.5 |
|
| 0.8.4 |
|
| 0.8.3 |
|
| 0.8.2 |
|
| 0.8.1 |
|
| 0.8.0 | First release. |
To contribute to this project, please take a look at the Contributing Guidelines first. Please note that this project is released with a Code of Conduct. By contributing to this project, you agree to abide by its terms.