Home
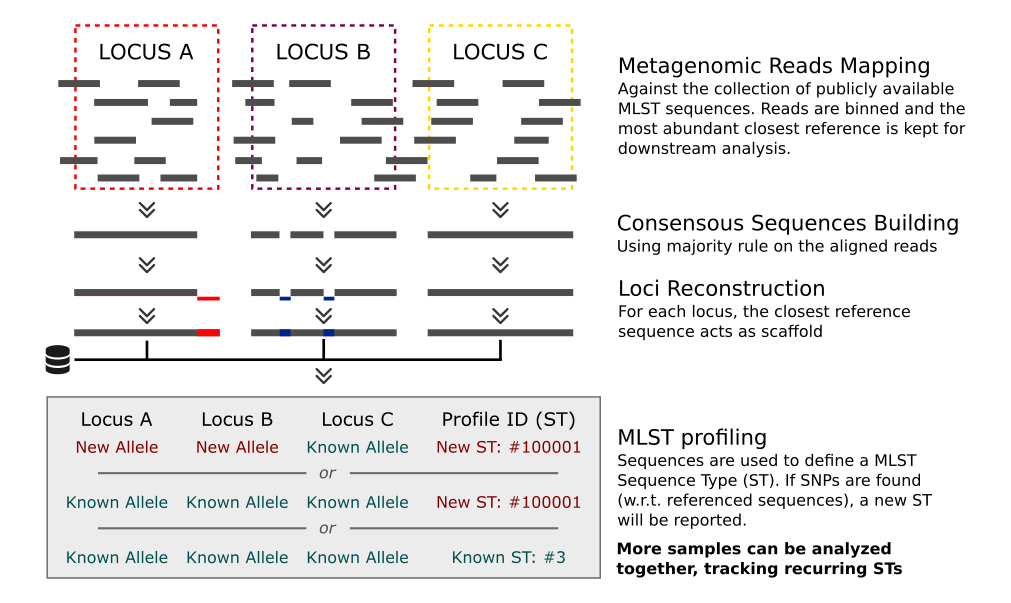
MetaMLST is a computational tool for strain level identification from metagenomic data. It exploits the Multi Locus Sequence Typing approach and performs and in-silico reconstruction of the MLST-specific loci.
To use MetaMLST you will need the following packages and tools:
- Bowtie2 >= v. 2.2.6
- Samtools >= 1.3.1
- Biopython >= 1.63
- Pysam >= 0.11.1
- Python3 (mlst.py requires Python3 >= 3.7)
MetaMLST takes as input Shotgun Metagenomic NGS Reads in FASTQ format (e.g. Illumina Hi-Seq). MetaMLST only works with shotgun sequencing data, and it is not applicable to 16S rRNA sequencing datasets.
MetaMLST outputs (by default in ./out):
- a list of detected MLST-trackable microbial species.
- A tab-separated file containing the typings of each sample provided. One file for each species.
- A tab-separated file containing the updated typing table (i.e. known and newly identified Sequence Types). One file for each species.
- A FASTA or CSV file containing the sequences of the MLST-reconstructed loci for each sample. One file for each species. (see the
--outseqformat option)
Note: MetaMLST can identify new loci-sequences (i.e. sequences different to any other sequence in the database) or new STs (i.e. novel combinations of loci). Those are labelled with a progressive number greater than 100000 (i.e. ST 100001 will be your first novel ST).
The MetaMLST consists of four phases:
- Retrieval of the available MLST data and creation of the MetaMLST-db ▸ metamlst-index.py;
- This step can be skipped if you use the pre-computed database (metamlstDB_2021.db), which will be downloaded automatically if you do not specify any custom db.
- Mapping of the metagenomic reads against the retrieved reference sequences ▸ (Bowtie2);
- Detection of microbial targets and reconstruction of the sample-specific MLST loci ▸ metamlst.py;
- ST calling and downstream comparative analysis ▸ metamlst-merge.py
- metamlst-index.py (index- and DB- building, Step 1)
- metamlst.py (Aligned Samples processing, Step 3)
- metamlst-merge.py (Multi-Samples merger, Step 4)
Step 0: Make sure you have all the requirements installed and available.
Step 1: Clone the repository (or download and extract the full repository from https://github.com/SegataLab/metamlst/):
git clone --recurse-submodules https://github.com/SegataLab/metamlst.git
cd metamlst
Step 2: Create a Bowtie2 index from the default MetaMLST database.
metamlst-index.py -i bowtie_index
Step 3: Use the index to map your FASTQ file(s):
bowtie2 --very-sensitive-local -a --no-unal -x bowtie_index -U YOUR_READS.FASTQ | samtools view -bS - > YOUR_ALIGNMENTS.bam
Step 4: Run MetaMLST on the BAM file. The results will be saved in ./out:
metamlst.py YOUR_ALIGNMENTS.bam
[Repeat Step 3-4 for each sample of interest]
Step 5: Run MetaMLST-merge on the the metamlst.py output files. The results will be saved in ./out/merged:
metamlst-merge.py ./out
Check out the Examples Section for practical examples on how to use MetaMLST: You can also download some MetaMLST Examples Scripts.
- Pubmlst.org
- PHYLOViZ: Minimum Spanning Trees generation and visualization
- Archaeopteryx: Phylogenetic Trees viewer
- MetaMLST Users Support Group
- Write an email to moreno.zolfo [at] unitn.it